Chapter 2: Abnormal Circulation
Now that we have amalgamated the ten unique characteristics into an integrated concept of normal cardiovascular function, we proceed to examine the implications of those ten unique characteristics during pathological states.
Heart Failure (Pump Energy Failure) Contrasted
with Pump Energy Excess
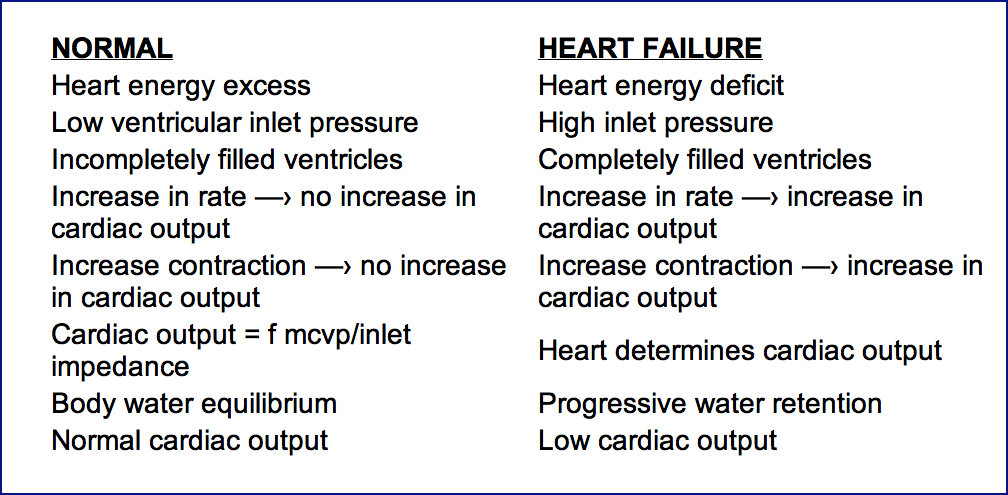
Heart failure is the only situation where the non-sucking heart determines its output. In this state, the heart is pumping at its maximum output and therefore, in effect, by limiting the output, it determines the output.
Normally, the heart rate and pump energy are in excess of that needed to eject enough blood at systole, so that the ventricles are empty enough at diastole to allow unobstructed passive filling. Normally ventricles are not maximally filled so there is reserve compliance which allows cardiac output to be determined by the extra-cardiac factors, rather than being limited by the heart. If the heart fails, it pumps less than the normally controlling extra-cardiac factors would dictate. During pump energy failure the heart's output is being limited by whatever amount it is able to pump. The heart in such a situation of failure, by limiting output, becomes the sole regulator of cardiac output.
Definition: Heart failure exists whenever heart function is inadequate to produce the output which would be allowed by the mean cardiovascular pressure and inlet impedance. Heart failure is present when cardiac function is limiting and, therefore, determining cardiac output. Normally, extra-cardiac factors being constant, if the heart contracts more strongly, efficiently, or rapidly, no increase in output occurs. Normally, the heart is exerting excess energy over that used to produce cardiac output and thus a reserve is present, so that increases in circulation rate are not impeded by the heart. Normally, at a given circulation rate, an increase in heart rate or increase in strength of cardiac contraction does not increase the output. However, during failure where the heart is pumping out the maximal circulation rate that it can produce at the moment, increases in the rate and strength of contraction do increase cardiac output, up until venous pressure is reduced to normal, at which time the heart is no longer in failure.
When heart failure occurs, blood volume equilibrium shifts to the system behind the failing ventricle with an increase in the venous pressure at the inlet of the failing ventricle. The other ventricle, in the presence of the slowed circulation rate, is able to maintain a low inlet pressure, with the resulting shift in blood volume equilibrium toward the circuit behind the failing ventricle. With left ventricular failure, for instance, the circulation slows to the rate limited by the left ventricle and a new equilibrium occurs with a shift of blood volume from the systemic circuit to the pulmonary vascular bed. Pulmonary edema occurs if this shift in equilibrium is severe.
The high end-diastolic ventricular pressure and high end systolic volume, from a low ejection fraction in heart failure, would also be factors limiting flow, if the heart weren't already determining a lower output.
A secondary change occurs when the left ventricle fails. The lowered cardiac output decreases renal blood flow, thereby causing retention of fluid and elevation of the mean-vascular pressure. Fluid equilibrium, which exists between the cardiovascular system and extra-vascular spaces, then results in edema, liver engorgement, and the other fluid abnormalities of congestive heart failure. Heart failure can intensify in a descending spiral if the blood volume progressively expands. The over-stretching of the heart puts it at a poor mechanical advantage, thus further increasing its failure (the descending limb of Starling's curve).
An extreme example of right heart failure is seen after a "Glen" right atrial-pulmonary artery anastomosis procedure for palliation of tricuspid atresia. Here, the person has virtually complete failure of a non-functioning right heart, with a surgical bypass of the right ventricle so that, in effect, the circulation is by the left ventricle alone. Circulation is slowed by the increase in impediment that normally would be overcome by the right ventricle. As a consequence of the slowed circulation, the kidneys receive slow flow, causing retention of fluid, resulting in a permanent elevation in the homeostatic level of mean cardiovascular pressure, resulting in some compensatory increase in cardiac output at the new equilibrium.
Causes of Heart Failure:
Cardiac causes (low myocardial power):
- Myocardiopathies (viral, arteriosclerotic, toxic, rheumatic)
- Incompetent leaky valves, e.g., mitral or aortic
- Abnormally slow heart rate, e.g., complete heart block
Peripheral vascular causes:
- Abnormally high mean cardiovascular pressure causing potentially higher circulation rate than a normal heart is able to deliver, e.g., renal shutdown resulting in high output failure
- Abnormally low impedance to the heart, e.g., arteriovenous fistula
Signs and Characteristics of Heart Failure:
- High venous pressure is the major pathognomonic finding in cardiac failure. If the heart were not in failure, the pressure would be normally low.
- A new equilibrium occurs between the two vascular systems with a shift in volume to the field behind the failing ventricle. I.e., left ventricular failure shifts the equilibrium with a greater volume in the pulmonary circuit, resulting in x-ray evidence of pulmonary vascular engorgement and high pulmonary venous pressure. With right heart failure, the neck vein pressure is seen to be elevated. However, the systemic system initially is large enough to accommodate the shift in blood volume from the lungs without significant venous engorgement until secondary changes (as seen below) occur.
- With either left or right heart failure, increased fluid accumulation in the body progresses in a descending spiral caused by slowed cardiac output → less than optimum renal blood flow → low renal output → retained fluid in the vascular system → increased mean cardiovascular pressure → edema → liver engorgement and ascites.
Pathologic Conditions Resulting in Heart Failure:
- Complete heart block with slow pulse
- Extremely rapid pulse
- Intermittent, pulsatile venous flow
- Low ejection fraction
- Myocardiopathy with weak myocardial contraction
- Myocardial infarction to the point of low ejection volume
- Valvular heart disease with stenosis or insufficiency
- Myocardial failure
- Pericardial tamponade
- Constrictive pericarditis
Goals in the Treatment of Heart Failure:
- Make the heart contract stronger and faster.
- Lower the resistance to ejection of blood from the ventricles.
- Lower the mean cardiovascular pressure by salt and water restriction and diuretics
- Restore the atrial effect if it is compromised by atrial fibrillation or nodal rhythym.
If failure is due to myocardial contractility, lessening the failure state by lowering mean cardiovascular pressure to the point of producing normal venous pressure, without increasing myocardial energy, may create hypovolemic shock rather than restoring normal circulation.
Shock
Shock is a low cardiac output state that results from low mean cardiovascular pressure. The low mean cardiovascular pressure can result from altering of one or the other of the two determinants of that pressure: (1) Increasing vascular compliance, and (2) Loss of blood volume.
- Low output with resulting hypotension from sudden loss of vascular tone is frequently seen after inducing spinal anesthetics. A dramatic example was seen following total central nervous system anesthesia, induced when marcaine was inadvertently injected into the spinal canal during an attempt to do an intercostal nerve block. In this case, sudden total relaxation of the entire vascular system resulted in severe hypotension. Loss of vascular tone can be reversed by the use of vasopressors and the addition of fluid.
- Shock from loss of blood volume can be temporarily compensated for by decreasing vascular compliance by vasopressor drugs, but ultimately needs to be corrected by blood volume restoration.
A combination of the two mechanisms of shock is seen in anaphylaxis, where there is both relaxation of the vascular system plus a shift in blood volume to the extra-vascular space, caused by increased capillary porosity.
Whatever the cause, the resulting low mean cardiovascular pressure in shock can be restored to normal by rapid infusion of fluid and electrolytes, aided by the use of vasopressor drugs.
During both of the shock mechanisms, the heart is already spending excess energy, so increasing the heart rate or strength of contraction will not increase the cardiac output. The obvious increase in output that follows the use of epinephrine, or other vasopressors, in such states is not caused by the inotropic effect on the heart, but by their effect of increasing the mean cardiovascular pressure by decreasing vascular compliance. Thus another temporary therapeutic measure, which increases mean cardiovascular pressure by making the system less compliant, is the surrounding of the vascular system by a "G-suit" such as those used by emergency medical personnel. It matters little how fast or how strongly the heart contracts: If there is no pressure in the cardiovascular system, there can be no circulation.
Arterial Hypertension (Low and High Output Types)
Arterial hypertension results from two radically different mechanisms. One is caused by increased arteriolar resistance in a system with normal mean cardiovascular pressure. The other results, in the presence of normal arteriolar resistance, from elevated mean cardiovascular pressure.
- In arterial hypertension from increased arteriolar resistance, there is no slowing of the circulation rate from the resistance. The heart, which normally expends excess energy, just forces whatever blood that comes to it right past the resistance point. The only change in circulation is the elevated arterial pressure. Such arterial abnormal resistance may have its origin from transient neuro-humeral stimuli. However, with longstanding arterial hypertension, changes eventually occur, resulting in more permanent structural resistance increase. If, eventually, the vascular changes effect blood flow to the kidneys, the second type of hypertension may develop.
- High mean cardiovascular pressure hypertension: The most graphic example of this state is seen during renal shutdown, from whatever cause. Here, if fluid and electrolyte intake continues, and there is reduced fluid leaving the body, the vascular volume progressively rises, with a corresponding increase in mean cardiovascular pressure. If the heart is strong enough that it continues to exert excess energy over that necessary to maintain a low ventricular inlet pressure, cardiac output increases. With the increased flow, even in the presence of unaltered arteriolar state, the resulting arterial pressure is elevated. If the heart is normal, but the pressure becomes so high that venous pressure rises, high output heart failure occurs. Severe high output failure has been seen where the systemic arterial pressure was 350/200, the systemic venous pressure was elevated with the neck veins at 30 cm., the pulmonary artery pressure was doubled, and the pulmonary capillary pressure was so high as to cause pulmonary edema with frothing of blood from the mouth and nose. This is in marked contrast to the distribution hypertension caused by arteriolar resistance increase.
The spectrum of hypertension states varies greatly between these two extremes and combinations of the two. Determination of the mean cardiovascular pressure helps delineate the significance of each in diagnosis. (See the Appendix on the clinical determination of mean cardiovascular pressure.)
Therapy for the two types of hypertension should be aimed at the etiology. For high mean cardiovascular hypertension, improving renal blood flow by removing renal arterial obstruction, diuretics, low salt diet, and water restriction to lower mean cardiovascular pressure or even renal transplant may be indicated. For hypertension caused by high arteriolar resistance, arteriolar vaso-relaxing drugs and changing the patient's response to stress may be effective.
Arteriovenous Fistula
An arteriovenous fistula, with blood flow going directly from an artery to a vein without going through arterioles, capillaries, and venules, bypasses most of the resistance that has upstream compliance. Therefore, depending on size, a fistula can cause tremendous increase in cardiac output. If a shunt is great enough, high output failure can result. If a shunt is in an extremity, where it can be occluded by manual pressure, alternating decrease and increase of cardiac output can be observed, as the shunt is intermittently occluded and opened. This maneuver is a good demonstration of the peripheral vascular control of circulation rate.
Arteriovenous fistula shunts occur in a number of disease states. For example, blood flow is shunted from arteries to veins in thyrotoxic goiters. Rather than there being one large arteriovenous channel, there are many small shunting vessels. The blood may be flowing so rapidly through the many small shunts that a loud, continuous murmur may be heard over the thyroid gland. This arteriovenous shunting of blood flow accounts, in part, for the high cardiac output associated with toxic goiter.
Changes In Size of the Vascular System
Because of the peripheral control of cardiac output, changing the size of the vascular bed, permanently (such as by a leg amputation) or by short term decrease (such as by cross-clamping the aorta and vena cava during surgery), causes an instantaneous decrease in cardiac output, mechanically, without any involvement or need of neurologic or humeral reflexes.
Complete Heart Block and Fixed-Rate Pacemakers
Patients with heart block, with a very slow pulse rate, have maximal ventricular filling before the completion of diastole, thereby restricting venous flow. This restriction in venous flow causes high venous pressure and low cardiac output. As the rate is incrementally increased by the use of a pacemaker, pressure gradually falls, until the ventricles are no longer maximally filled at diastole. Further increase in pacemaker rate above that level causes no more increase in output and no further drop in the venous pressure, as ventricular capacity is no longer restrictive to flow. A rate of 60 produces this plateau in most adults at rest and in a recumbent position. If the patient is active, the pacemaker rate may need to go to 80 or even 100 before further increases in pacemaker rate cause no corresponding increase in cardiac output. When the rate is fixed at 80 and the patient is resting, the output goes down normally with a waste of energy expended by the heart, the excess not being used for circulation. The rate of 80 allows a great variation in activity without exceeding a need for pulse increase to prevent failure. If the pacemaker is set at 120, such a rate would probably accommodate cardiac output for any violent activity. However, at rest, even though the output goes down to normal, the 120 pulse tachycardia is uncomfortable, the excess myocardial energy waste is great, and myocardial oxygen availability may be taxed. Therefore, fixed pacemaker rates are usually set at about 76 to 80 as a compromise between excess myocardial energy spent for circulation at rest and needed cardiac energy for strenuous activity.
From pacemaker experience we conclude that: (1) The heart, when not in failure, is always expending excess energy over that necessary to produce cardiac output. (2) The ability to lower pulse rate below maximum during periods of low cardiac output has implications for energy conservation rather than cardiac output.
Left Ventricular Booster Pumping
Myocardial energy failure is a frequent finding during acute myocardial infarction and for short periods of time after coming off of bypass after open heart surgery. In both of these situations, left ventricular booster pumping has proved useful during a recovery period.
A competent, passive filling, pulsatile outflow, continuous inflow assist pump, placed between the left atrium and aorta will automatically lower the left atrial pressure and restore circulation rate to normal. A passive filling booster pump, which expends excess energy, put in parallel with the failing left ventricle, will make the combined output of the two normal. The combined output of two non-sucking pumps in parallel will act as one pump which produces excess to that needed for normal cardiac output. Their combined output will be controlled by the extra-cardiac factors that normally control circulation rate. Whatever amount the ventricle is unable to pump will run automatically into the booster ventricle and be ejected into the aorta. When the heart has recovered, the left atrial and arterial blood pressure will remain the same whether the pump is on or off. One method of weaning the patient off of the booster pumping is to raise the pump a few centimeters above the left atrium. Blood will then run preferentially into the heart's ventricle and be pumped out by it, if the heart is no longer in power failure. If the ventricle is still in failure, the venous pressure will rise only those few centimeters. Then whatever the heart does not pump the booster pump will.
From cardiac booster pumping, we conclude that the non-failing heart is expending excess energy to that needed at any moment to produce its output. Expenditure of energy by a parallel passive filling booster pump cannot increase the circulation rate above that dictated by extra-cardiac determinants.
SUMMARY: The cardiovascular system is a closed elastic circle, containing two passive filling pumps in series with two vascular beds, systemic and pulmonary. Normally, the circulation rate made by the two pumps is controlled by mean cardiovascular pressure and inlet impedance. It is only during heart failure, when heart function is limiting the cardiac output, that the heart is regulating circulation rate.